
What is Rowan? Rowan is a cloud-native molecular-modeling platform that runs physics-based and machine-learning chemistry workflows (pKa, logD, tautomers, solubility, permeability, docking, MD, and more) through a scientist-friendly web app and a Python API. The skill benchmarked here was contributed by Rowan Scientific to K-Dense's open-source Scientific Agent Skills library.
Every computational chemist has RDKit open in a terminal. It is free, fast, local, and genuinely excellent at what it was built for: molecular I/O, substructure search, fingerprints, 2D descriptors, conformer geometries, tautomer enumeration. For a huge fraction of day-to-day cheminformatics, RDKit is all you need.
But RDKit is, by design, a cheminformatics toolkit, not a physics engine. Ask it for the pKa of a molecule and it has no answer. Ask for the logD at pH 7.4 that actually governs absorption, and it can only hand you a pH-blind logP. Ask which tautomer dominates in water, with populations, or for a docked pose, or a permeability estimate, and you are out of scope.
Rowan covers that different layer, and it is now available as an agent skill that Rowan contributed to K-Dense's open-source Scientific Agent Skills library: an LLM agent (here, Claude) can drive the whole platform from natural language. So we asked the practical question, and answered it with numbers:
Moving from the free-tool baseline to the Rowan skill, what do you gain, how accurate is it against experiment, and what does it cost?
Every result below has three reference points: Rowan, the without-skill baseline (RDKit, or a competent functional-group heuristic where RDKit offers nothing), and experiment (literature values). All code, data, and the roughly 60 cloud workflows are reproducible. The full study used about 12 to 14 Rowan credits, or roughly $0.48 to $0.56 at Rowan's published pay-as-you-go rate of $0.04 per purchased credit (Rowan Credits FAQ).
Study 1: pKa, the clean kill
pKa determines how much of your molecule is charged at physiological pH, which cascades into solubility, permeability, and binding. It is also the single most glaring hole in the free toolbox: RDKit ships no pKa predictor. Without a dedicated tool you fall back on functional-group rules of thumb ("carboxylic acid ≈ 4.5, aliphatic amine ≈ 10.5") or pay for commercial software.
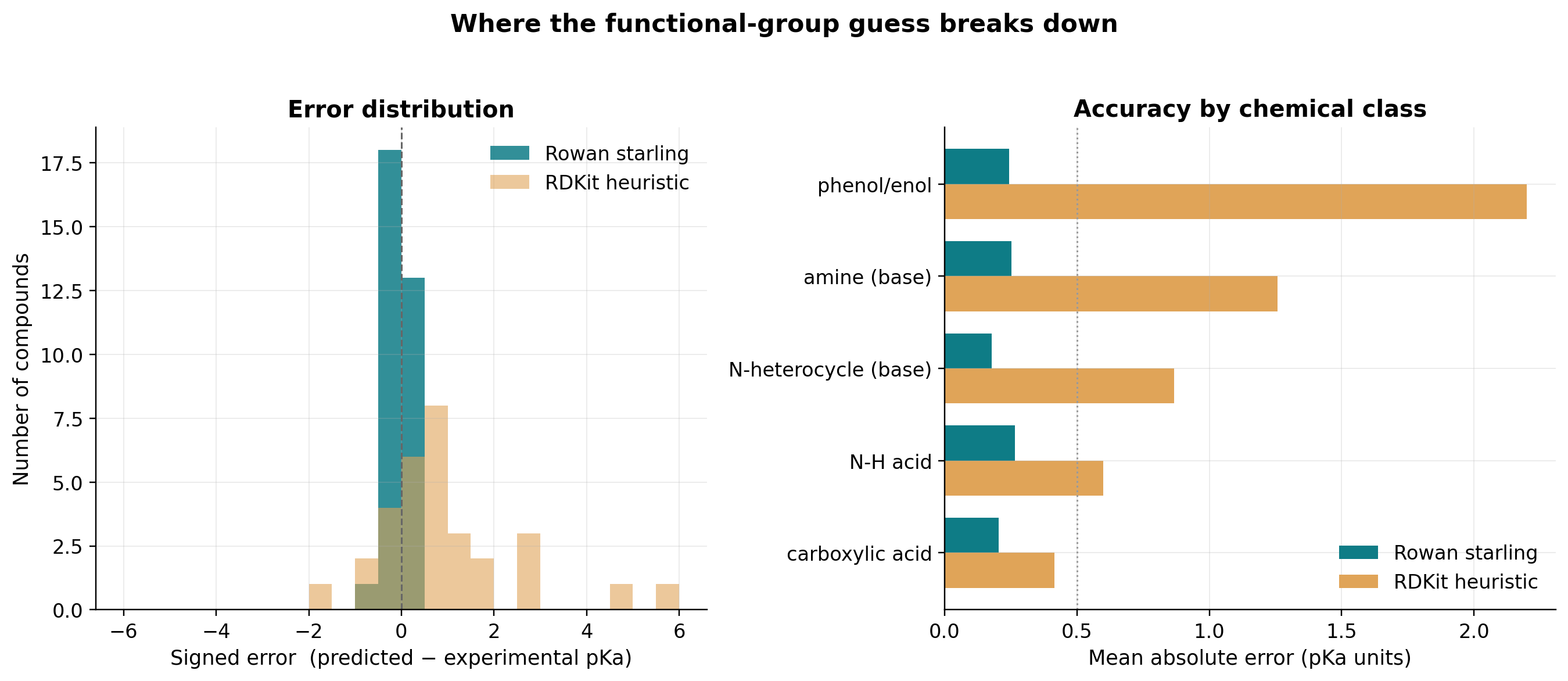
We took 32 drugs with reliable literature pKa (carboxylic acids, phenols, N-H acids, anilines, pyridines, imidazoles, and aliphatic amines) and compared Rowan's starling model against a faithful functional-group heuristic (the best you can do by eye).
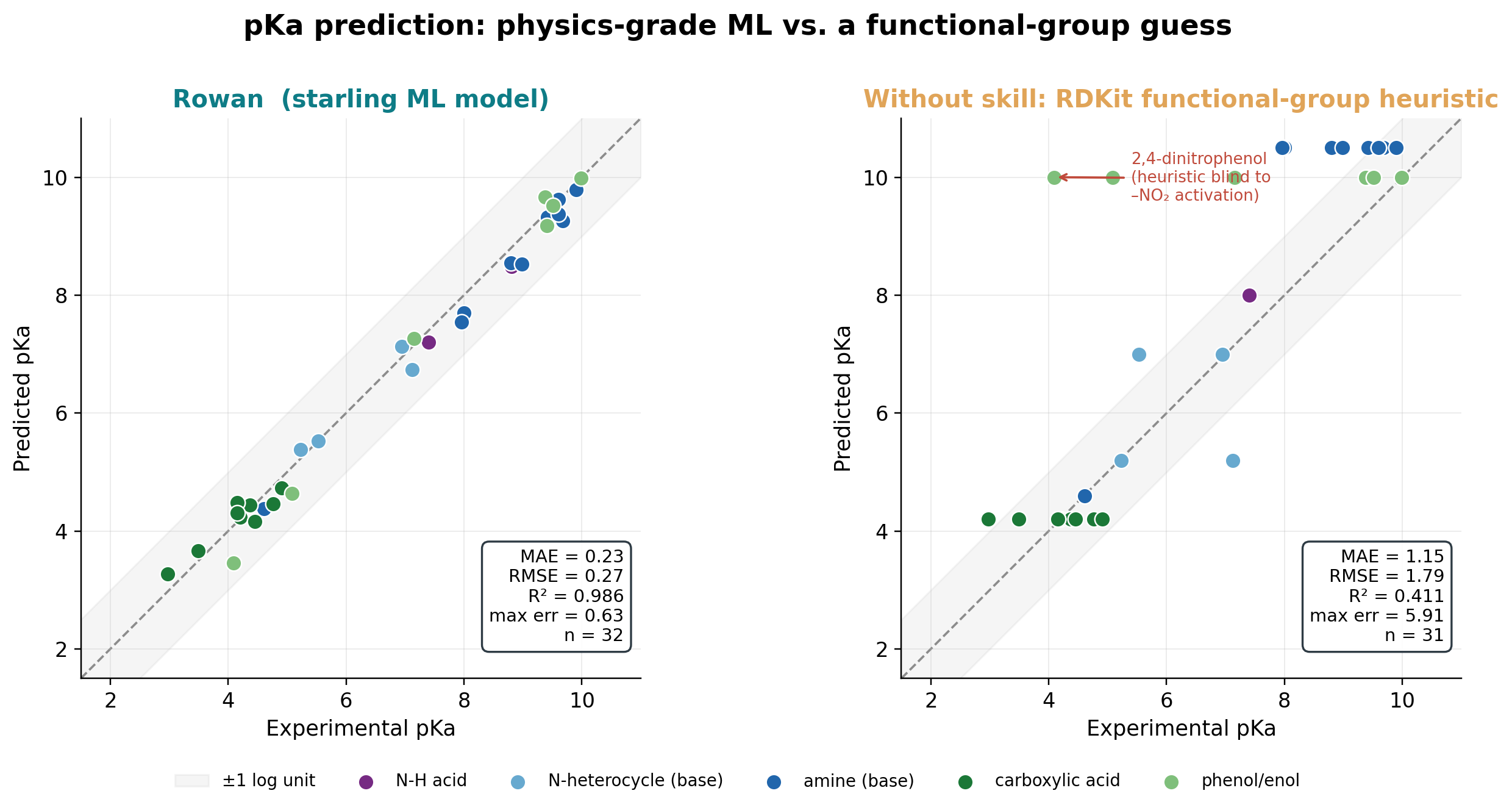
| Method | MAE | R² | max error |
|---|---|---|---|
Rowan starling |
0.23 | 0.986 | 0.63 |
| RDKit functional-group heuristic | 1.15 | 0.41 | 5.91 |
Rowan's mean absolute error of 0.23 pKa units sits at the level of experimental reproducibility. The heuristic, meanwhile, breaks down exactly where chemistry gets interesting:
- Substituent electronics. 2,4-dinitrophenol has an experimental pKa of 4.09; Rowan predicts 3.46; the heuristic says 10.0, because it cannot "see" that two nitro groups acidify the phenol by six log units.
- Amine individuality. The heuristic assigns every aliphatic amine 10.5. Rowan resolves nicotine (7.70), lidocaine (7.54), diphenhydramine (8.53), propranolol (9.32), and ephedrine (9.62), each within about 0.4 of experiment.
Field note for skill users. Rowan's pKa result exposes both strongest_acid and strongest_base. Match them to the physical ionization: acids use strongest_acid, bases use strongest_base. For propranolol, strongest_acid returns 10.0 (a spurious amide-like N-H deprotonation) while strongest_base gives 9.28, the number you actually want.
Study 2: lipophilicity, the right number versus the wrong one
Here is a subtler trap, and a more important one. RDKit's Crippen MolLogP gives you the neutral logP, a single pH-independent number. But the quantity that controls oral absorption is logD at pH 7.4, and for an ionizable drug it can be 2 to 4 log units lower, because almost all of the molecule is charged at physiological pH.
We took 16 drugs with experimental logP and logD₇.₄ (including four neutral controls) and compared RDKit's logP, used as the ADME-relevant lipophilicity as it often is in practice, against Rowan's pH-aware logD₇.₄ from the macropKa workflow.
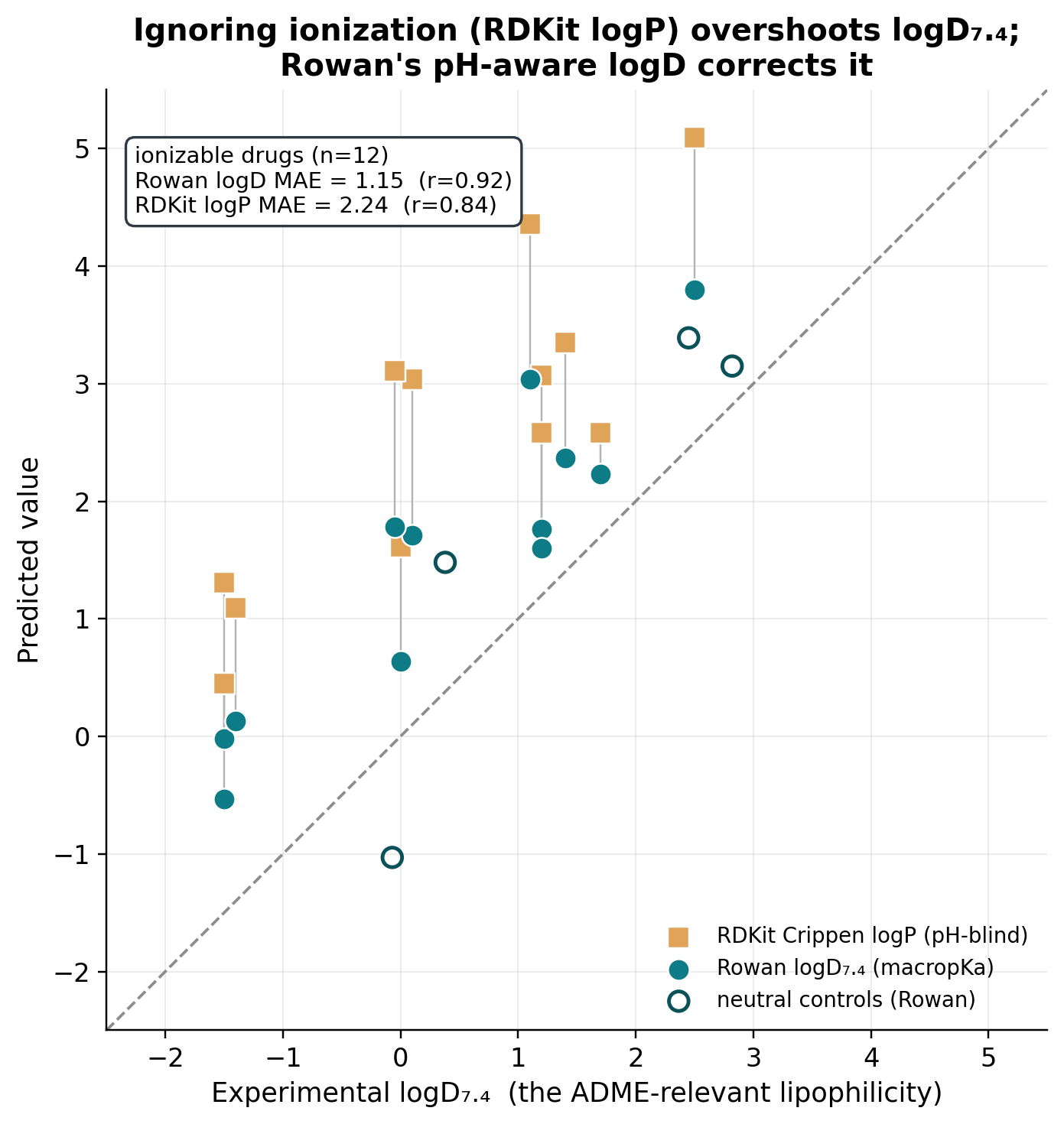
| Predicting experimental logD₇.₄ (12 ionizable drugs) | MAE | R² | r |
|---|---|---|---|
| RDKit Crippen logP (pH-blind) | 2.24 | −2.36 | 0.84 |
| Rowan logD₇.₄ | 1.15 | 0.04 | 0.92 |
RDKit's logP-as-logD has a negative R². For ionizable drugs it is literally worse than guessing the dataset average, because it systematically overshoots. Rowan halves the error, correlates strongly (r = 0.92), and gets the ionization direction right for all 12 compounds. On the four neutral controls (caffeine, antipyrine, diazepam), Rowan correctly applies zero correction: logD ≡ logP, exactly as it should.
But the real payoff is not a single number. It is the whole curve, which RDKit fundamentally cannot draw:
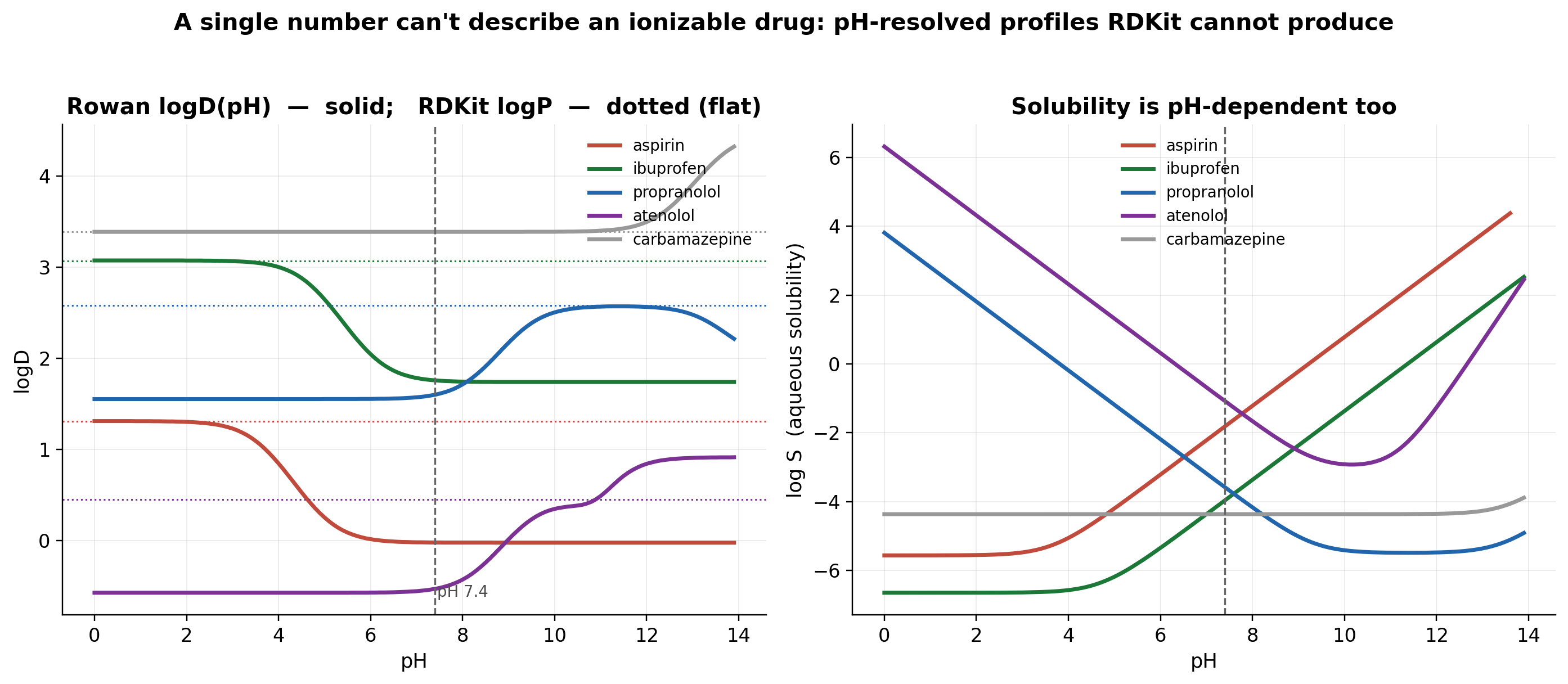
Acids shed lipophilicity above their pKa; bases below it; neutrals stay flat; and aqueous solubility swings the opposite way. This pH-resolved picture is what a formulation or ADME scientist actually reasons with.
Honest caveat. Rowan's neutral-logP plateau tracks RDKit's Crippen logP almost exactly. Rowan's value-add is the ionization correction layered on top, not the neutral logP itself. And its model lets carboxylate anions retain some octanol partitioning, so it under-penalizes acids by about 1 log unit. logD is a genuinely hard endpoint; trust the shape and the pKa, and treat absolute acid logD₇.₄ as mildly optimistic.
Study 3: tautomers, physics versus convention
Tautomers are where it pays to be skeptical. RDKit's TautomerEnumerator can enumerate tautomers and pick a canonical one, but that choice is a deterministic scoring convention (rules that favor amide over imidic acid, keto over enol, thione over thiol). It is perfect for registration and deduplication, but it carries no energies, no populations, and no awareness of solvent, temperature, or substituents.
Rowan's tautomer search optimizes and ranks tautomers by solvated free energy and returns ΔG and Boltzmann populations. We tested six classic systems with known aqueous-dominant forms.
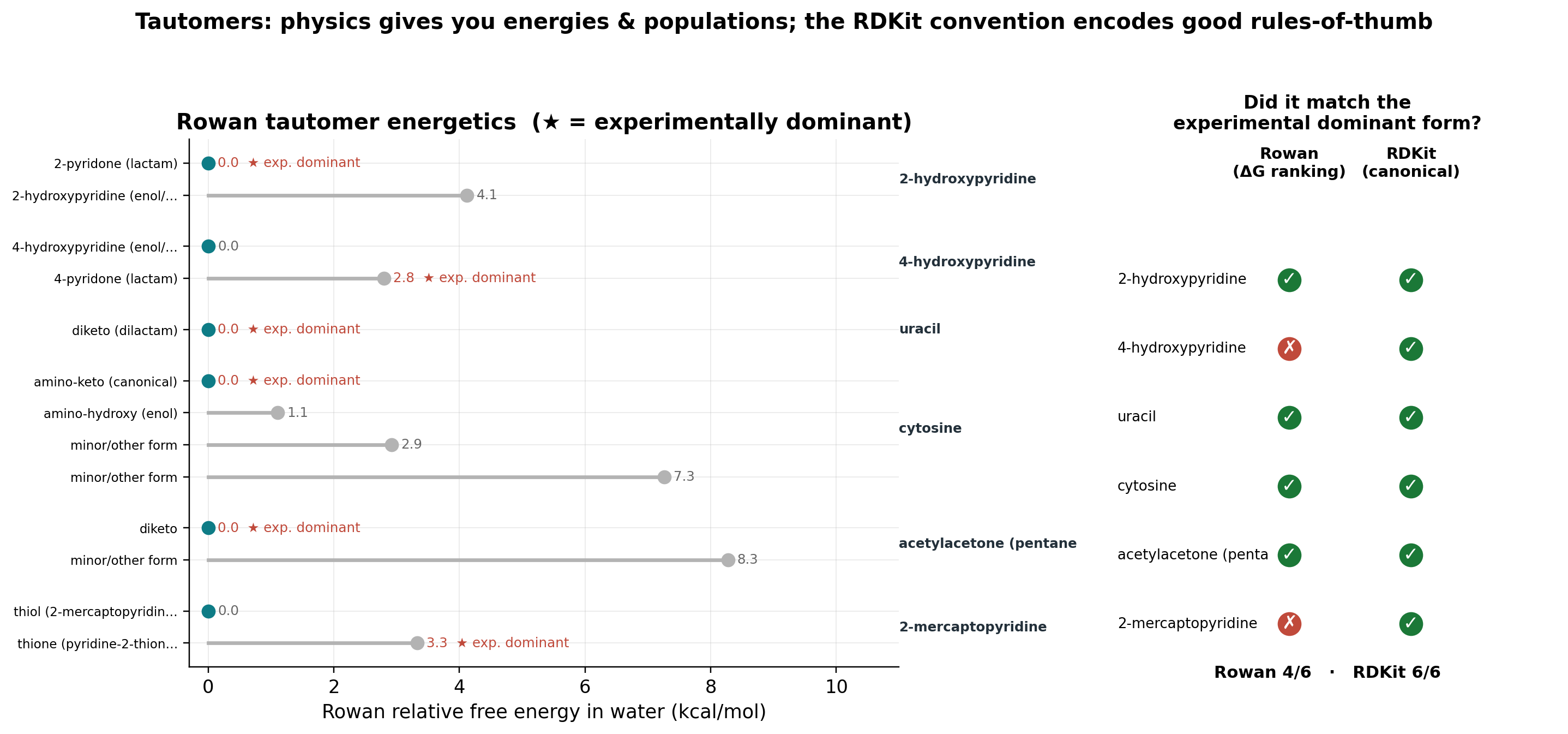
| System (aqueous dominant) | Rowan ΔG ranking | RDKit convention |
|---|---|---|
| 2-pyridone | ✓ (99.9%) | ✓ |
| 4-pyridone | ✗ | ✓ |
| uracil (diketo) | ✓ | ✓ |
| cytosine (amino-keto) | ✓ | ✓ |
| acetylacetone (diketo, aqueous) | ✓ | ✓ |
| 2-mercaptopyridine (thione) | ✗ | ✓ |
| Total | 4 / 6 | 6 / 6 |
The tautomer benchmark is useful precisely because it is mixed. Rowan nails the biologically canonical forms (2-pyridone, uracil, cytosine) but misranks the two equilibria most sensitive to explicit solvation (4-pyridone and 2-thiopyridine) because its default uses an implicit solvent model. RDKit's convention matches all six because its hard-coded rules encode those common motifs, but it would give the same answer in chloroform, and tells you nothing about how much of each form is present.
The lesson is "right tool for the job": if you need a canonical structure for a database, use RDKit; if you need ΔG and populations or substituent sensitivity, use a physics method, and validate the polar, solvation-sensitive cases.
(We also caught, and disclose, a ground-truth subtlety: acetylacetone's famous enol dominates in gas and neat liquid, but in water the diketo form wins about 85%. Since Rowan models the aqueous phase, the correct aqueous reference is diketo.)
Study 4: a full ADME profile, orchestrated end-to-end
The skill is not just individual predictions, it is the agent chaining them. From a SMILES string, the agent builds a complete profile: descriptors → macropKa (logD and solubility vs pH) → Caco-2 permeability. We ran three drugs that span the Biopharmaceutics Classification System.
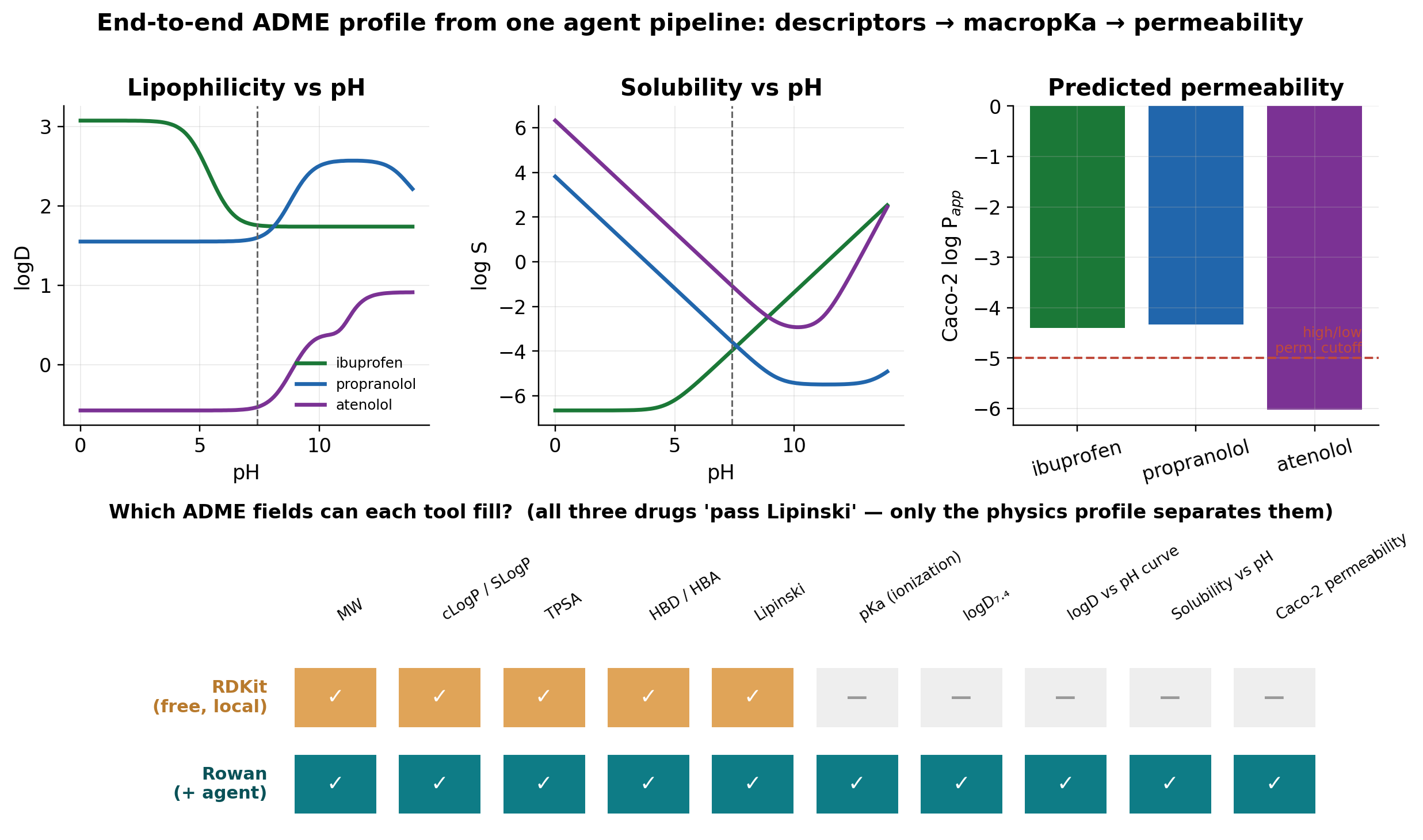
| Drug (BCS class) | logD₇.₄ | logS₇.₄ | Caco-2 logP_app | Lipinski |
|---|---|---|---|---|
| ibuprofen (II) | 1.76 | −3.97 (low sol) | −4.40 (high perm) | pass |
| propranolol (I) | 1.60 | −3.58 | −4.33 (high perm) | pass |
| atenolol (III) | −0.53 | −1.08 (high sol) | −6.03 (low perm) | pass |
The predictions reproduce textbook pharmacology: atenolol's notoriously poor passive permeability shows up as a Caco-2 logP_app nearly two log units below the others, while its high solubility and ibuprofen's low solubility match their BCS classes. The punchline: all three "pass Lipinski" in RDKit, so the free 2D filter cannot tell a BCS I drug from a BCS III drug.
Study 5: into 3D, docking pose recovery
Structure-based work is the last frontier RDKit does not touch. Rowan's docking is managed AutoDock Vina, so the comparison here is not accuracy-versus-RDKit, it is operational: with the skill it is a PDB id, a SMILES, and a pocket box in a few lines.
We validated it the standard way: redock benzamidine into bovine trypsin (PDB 3PTB) and measure the heavy-atom RMSD of the top pose against the crystallographic ligand. The success criterion is RMSD < 2.0 Å.
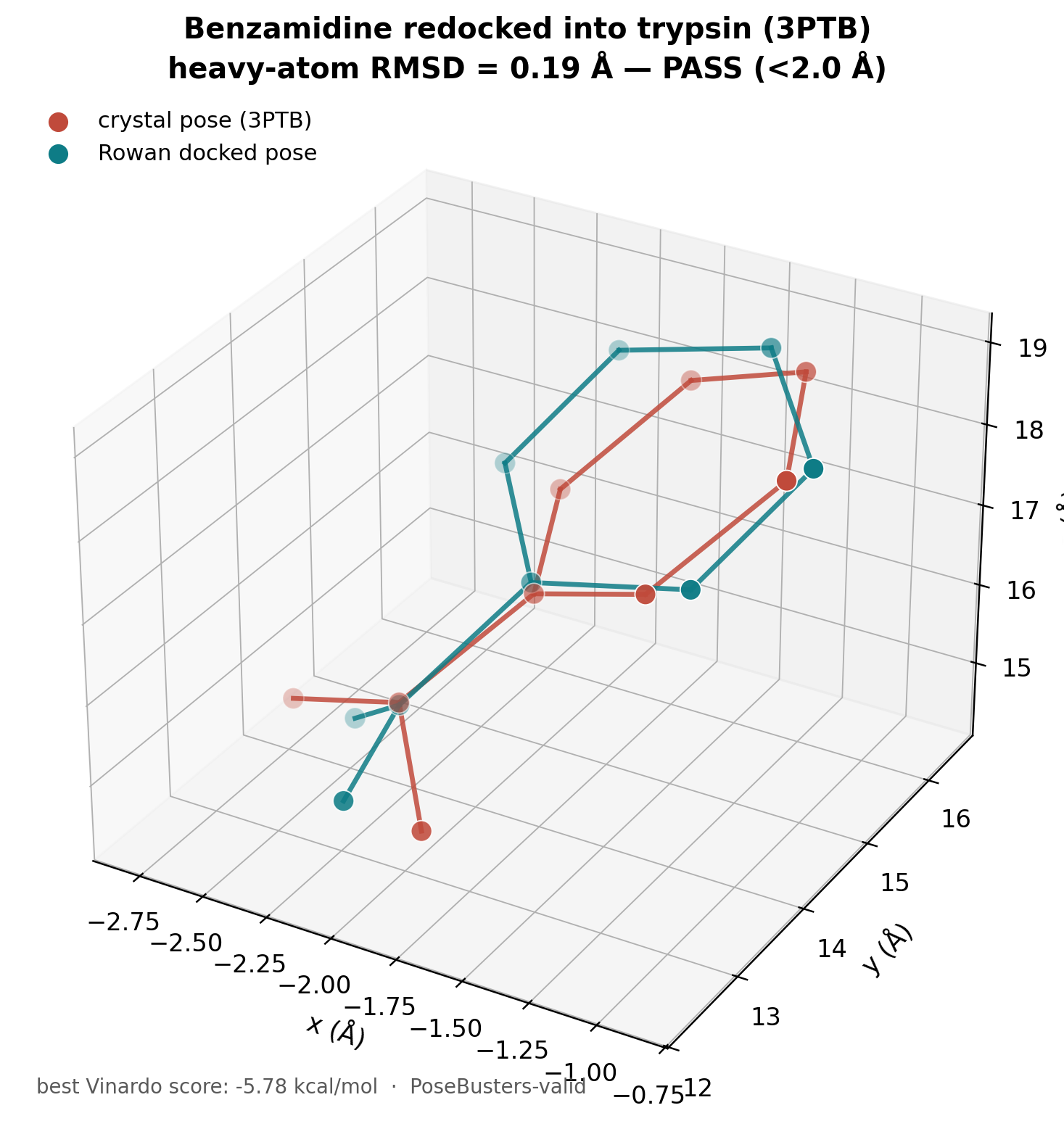
From a PDB id and a SMILES string, the run produced 13 PoseBusters-valid poses (best Vinardo score −5.78 kcal/mol), and the top pose reproduced the crystal binding mode to a heavy-atom RMSD of 0.19 Å, a textbook pass. The same result with a local toolchain would have meant installing Vina, preparing a PDBQT receptor, and hand-defining a grid box; here it was about 1 to 2 credits.
The big picture
The capability matrix below tells the whole story in one frame.
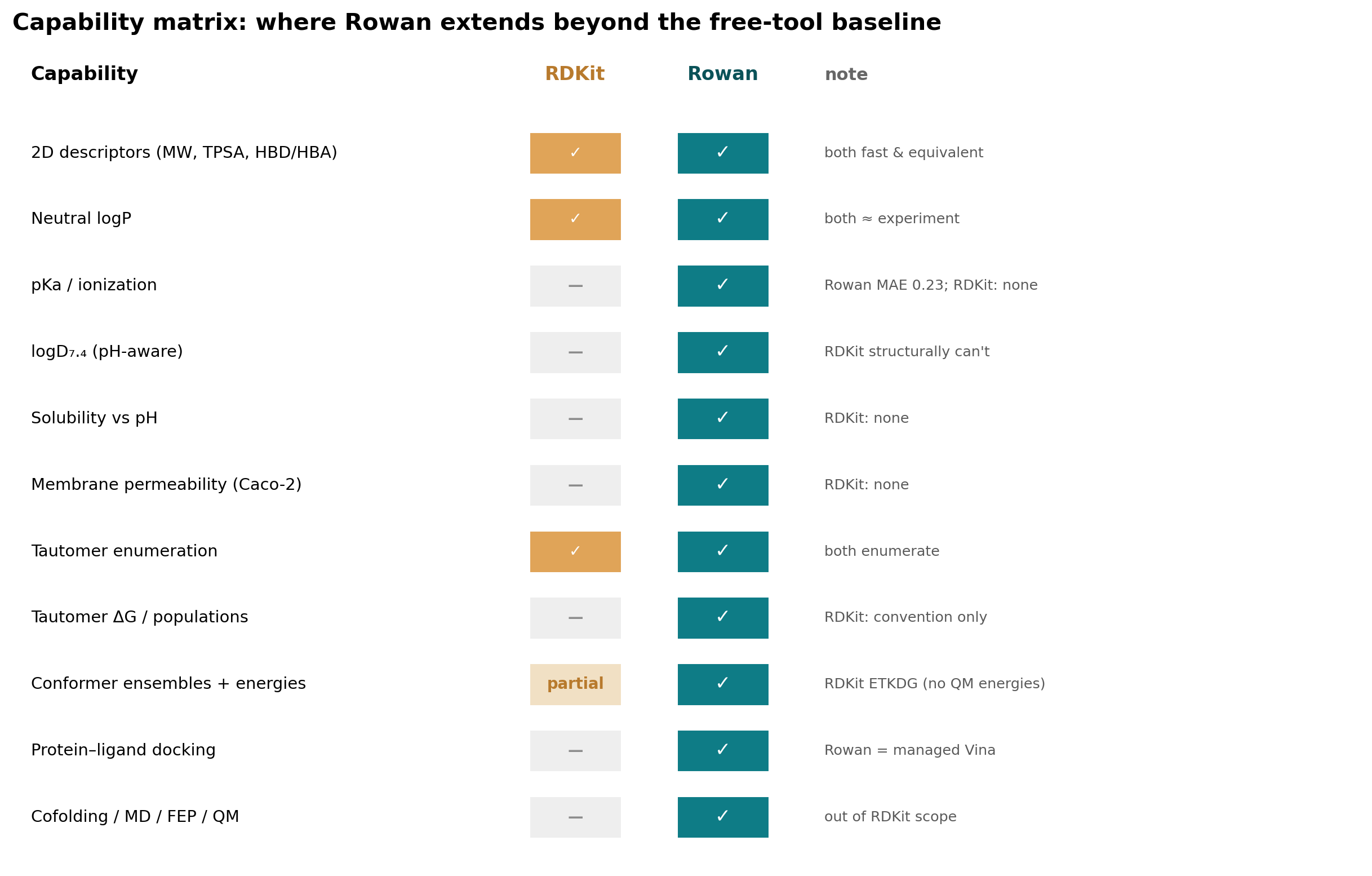
For the everyday work RDKit was built for (2D descriptors, neutral logP, enumeration), the free baseline is excellent and you should keep using it. But the moment a decision turns on ionization, pH-dependent behavior, solubility, permeability, tautomer energetics, or 3D structure, the Rowan skill moves the agent from descriptor calculation into chemistry workflows that RDKit was never meant to run.
| Property | Without skill | Rowan | vs. experiment |
|---|---|---|---|
| pKa | heuristic MAE 1.15; RDKit: none | MAE 0.23, R² 0.986 | at exp. reproducibility |
| logD₇.₄ (ionizable) | logP-as-logD MAE 2.24, R² < 0 | MAE 1.15, r 0.92 | right quantity and direction |
| logD / solubility vs pH | impossible | full curves | n/a |
| Permeability (Caco-2) | none | recovers BCS rank order | qualitative ✓ |
| Tautomer dominant form | 6/6 (no ΔG) | 4/6 + ΔG and populations | nuanced |
| Docking pose | DIY Vina pipeline | one call, managed Vina | RMSD 0.19 Å |
| Total cost | $0 / local | ≈ 13 credits, or ≈ $0.52 pay-as-you-go equivalent | n/a |
Keep RDKit for what it is great at. Reach for the skill when the answer depends on ionization, energetics, or 3D structure.
Reproducibility and methods
Every workflow result is checkpointed and keyed by molecule, so re-running the study re-collects cached results for zero additional credits. Baseline: RDKit 2026.03.2 (Crippen logP, TPSA, Lipinski, SMARTS matching, TautomerEnumerator, rdDetermineBonds). Rowan: rowan-python 3.x, called through the documented submit_*_workflow entry points. Ground-truth pKa, logP, and logD values are consensus literature numbers from the CRC Handbook, Avdeef's Absorption and Drug Development, and DrugBank-class compilations.
Going further: rowan-autosearch
The studies above score molecules one at a time. To put the same pieces into an optimization loop, see rowan-autosearch, an open-source harness for agent-driven molecular optimization. You define a chemistry objective and a starting molecule, and an AI coding agent acts as a medicinal chemist: proposing analogs, scoring them with Rowan's quantum and ML workflows, gating drug-likeness with RDKit, and iterating until a winning candidate emerges. Every candidate, Rowan payload, constraint check, and design rationale is logged as append-only JSON and rendered into an auditable HTML report. It is joint work between K-Dense and Rowan Scientific.
Get the skill
The rowan skill benchmarked here was contributed by Rowan Scientific to K-Dense's open-source Scientific Agent Skills library; you can read its full instructions in the rowan skill folder. To learn more about the platform it drives, visit Rowan.